

Advanced phase myeloproliferative neoplasms (AP-MPN) are associated with a very poor prognosis. The Phase Ib PHAZAR study set out to assess the safety & tolerability of oral ruxolitinib (RUX) in combination with 5-azaciditine (AZA) in patients (pts) with advanced-phase-MPN, defined as blast count >10%. The study included an observational arm for pts not suitable for the trial intervention. The clinical results of this study are presented in a separate abstract. Here we evaluate the molecular characteristics of PHAZAR pts and correlate with clinical features, outcome and therapy response.
Driver mutation (JAK2/CALR/MPL) allele burdens were quantified using targeted next-generation sequencing (NGS) and non-driver mutation analysis was performed using an ISO accredited Illumina TruSeq Custom Amplicon Panel, including 32 gene mutation hotspots & exons (~36,000 bp, 287 amplicons). SNP karyotyping was performed using the Illumina InfiniumOmniExpress-24v1-3 BeadChip assay. Data analysis was performed using R v4.0.
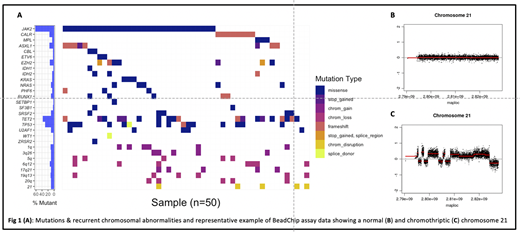
Clinical data were censored in February 2020, and NGS sequencing data were available for 24 interventional trial and 13 observational cohort participants. 11/13 observational pts received best supportive care, while 2/13 were treated with high-dose chemotherapy. All pts had a mutation in ≥1 targeted gene. 16% of pts were 'triple-negative' for MPN driver mutations, while 59%, 16% & 8% carried canonical mutations in JAK2, CALR & MPL respectively. 89% carried additional non-driver mutations, with a median of 2 (range 0-4) detected per pt (Fig 1A). Mutations in epigenetic regulators were detected in 21/37 pts (57%) (TET2, 38%; EZH2, 19%; ASXL1, 14%; PHF6, 5%; SETBP1, 3%) while 8/37 (22%) carried mutually exclusive spliceosomal mutations (SRSF2, 8%; U2AF1, 8%; SF3B1, 5%). 10/37 (27%) were TP53 mutant. High molecular risk (HMR) mutations (ASXL1, EZH2, IDH1/2, SRSF2, TP53, U2AF1 Q157) were detected in 24/37 (65%), and >1 HMR mutation in 7/37 (19%).
SNP karyotyping data were available for 42 pts (n=29 interventional, n=13 observational). 4/42 (10%) were wild-type, while 90% harboured >=1 chromosomal aberrations (median 4, range 0-16). Of these, 21 were recurrent in 3+ samples. 9 frequently recurrent events in >=5 samples included gains at 1q, 3q26, 17q21and losses of 5q, 6q12, 17p13, 19q13, 20q, and multiple losses and gains on chromosome 21q. 5 pts demonstrated evidence of chromothripsis. The presence of TP53 mutation was associated with a higher number of chromosomal aberrations (median of 3 vs 6.5, p=0.02).
Concerning clinical correlation, baseline driver mutation status did not impact on OS nor likelihood of achieving a durable response (DR, defined as having achieved a minimum of 6 months of complete or partial remission or stable disease as per published criteria (Cheson Blood 2006, Mascarenhas Leuk Res 2012)). The presence of >=3 additional mutations significantly impaired OS regardless of trial arm (1 yr OS 12% vs 55%, p=0.02), as did the presence of HMR mutations (1 yr OS 22% vs 73%, p=0.008) and TP53 mutations in isolation (1 yr OS 13% vs 55%, p=0.05). The presence of HMR mutations reduced the likelihood of achieving a DR (p=0.02). Pts with losses of >=1 chromosomal arms (other than 5q-) had a poor prognosis (1yr OS 27% vs 58%, p=0.05), while no pt with chromothripsis (n=5) survived to a year (1yr OS 0% vs 53%, p=0.002).
Mutational profiling of serial samples on therapy were available for 5 pts who achieved a remission during AZA and RUX therapy. One pt achieved a CMR but developed clonal evolution and emergence of a new ETV6 mutant clone at relapse. The other 4 cases demonstrated no change in clonal abundance during remission. This supports the hypothesis that response to AZA is mediated by alteration of subclonal contributions or prevention of further clonal evolution, rather than elimination of founder clones.
AP-MPN continues to confer a very poor prognosis and more effective therapies are urgently required. Genetic and molecular profiling of this prospective trial cohort demonstrates the high mutational burden and structural variants seen in this disease. Initial serial sample profiling demonstrates that molecular responses to AZA and RUX are rare and, where they occur, are not sustained. Incorporation of molecular profiling into trial design may help inform which patients are more likely to benefit from the intervention - e.g. those without evidence of chromothripsis at trial entry.
Harrison:Gilead Sciences: Honoraria, Speakers Bureau; CTI Biopharma Corp: Honoraria, Speakers Bureau; Celgene: Honoraria, Research Funding, Speakers Bureau; Janssen: Speakers Bureau; Incyte Corporation: Speakers Bureau; Novartis: Honoraria, Research Funding, Speakers Bureau; Shire: Honoraria, Speakers Bureau; AOP Orphan Pharmaceuticals: Honoraria; Promedior: Honoraria; Roche: Honoraria; Sierra Oncology: Honoraria. Drummond:Pfizer: Consultancy, Membership on an entity's Board of Directors or advisory committees; Astellas: Membership on an entity's Board of Directors or advisory committees; Takeda: Membership on an entity's Board of Directors or advisory committees; Jazz: Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; Novartis: Consultancy, Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding, Speakers Bureau; Gilead: Membership on an entity's Board of Directors or advisory committees; Bristol Myers Squibb: Honoraria, Membership on an entity's Board of Directors or advisory committees, Speakers Bureau; Blueprint Medicine Corporation: Research Funding. Knapper:Novartis: Honoraria, Membership on an entity's Board of Directors or advisory committees, Research Funding. Mead:Gilead: Consultancy; CTI: Consultancy; Abbvie: Consultancy; Celgene/BMS: Consultancy, Honoraria, Other: travel, accommodations, expenses, Research Funding; Novartis: Consultancy, Honoraria, Other: travel, accommodations, expenses, Research Funding, Speakers Bureau.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal